

How and Why Atherosclerosis Happens
Whether you have diabetes, heart disease or dementia...atherosclerosis is there. An almost universal risk factor. But, why? Let's review the evidence that nobody is talking about.
Background
If you are active in the health space, atherosclerosis is the talk of the town. Rightfully so.
How it comes about…and, indeed the downstream consequences is up for debate. But, one thing is for certain. Where there is chronic illness, there is almost always…atherosclerosis.
Like most aspects of the human body, atherosclerosis is also misunderstood.
First, some definitions.
Atherosclerosis is a subset of arterio-sclerosis.
Arterio-sclerosis simply means stiffening of the arteries.
Athero- is Greek for “gruel” or “paste” and refers to “fatty plaque”
Thus, atherosclerosis is the stiffening of arteries in the presence of “fatty plaque” otherwise known as an atheroma.
There are some things you should understand about the terminology used that is already misleading.
First, atherosclerosis is not a disease in itself. Similar to hypertension, atherosclerosis is a consequence.
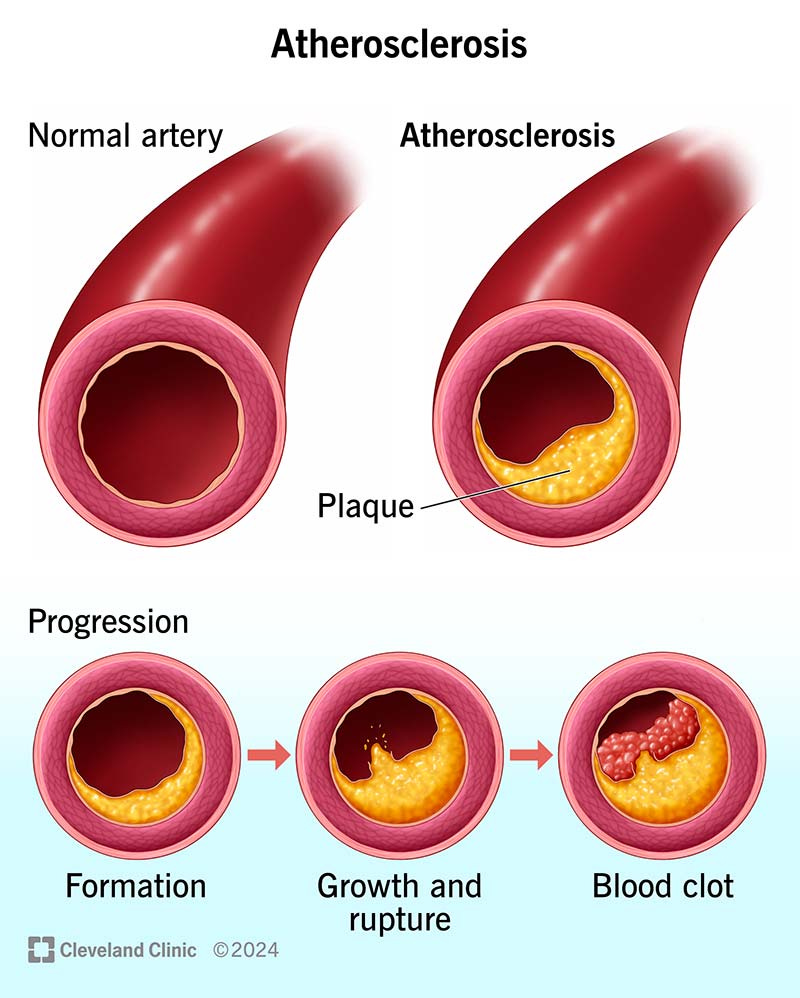
But, here’s the real kicker. Atherosclerotic plaques (atheromas), are almost entirely blood products, not fat. Not cholesterol. Not lipids.
I know what you are thinking, the above image from the Cleveland Clinic shows atherosclerosis as slow growing fat-deposition, which eventually gets clogged by a small blood clot.
Incorrect.
This is part of the marketing (so to speak), to get both the public and physicians to buy into the framework that athero-sclerosis is the result of fat deposition.
Even a simple Google or PubMed search will reveal the following about atheromas.
The primary component of these plaque is “fibrous” tissue, ranging from 50-87% depending on the plaque that is sampled
“Fibrous” tissue is yet another term that can be confusing
Fundamentally, this refers to breakdown and long-term clot evolution of blood products
In fact, the consequences of hemorrhage account for about 30-35% of the plaque’s volume
Another 1-15% of plaques are calcium/minerals
1-5% are inflammatory cells (yes, there is inflammation here…and we will soon reveal why that is important)
the Lipid (or fatty) core can range from 5-20% depending on plaque stability - another important concept
Furthermore, a lot of the lipids come from broken down cells (20 - 40%)
How much is left to be accounted for by circulating lipids? Very little.
An unstable lesion is one that is actively and rapidly changing. These are usually seen in patients with extremely poor metabolic health, very high inflammatory stress, and very poor vascular health overall.
Thus, it is no surprise that lipid content is higher in this population…as the body is vigorously trying to heal the damage being done to the blood vessel.
The Modern Discourse
If you keep up with the work of the most verbose and public figures in the realm of cardiovascular disease and “lipidology,” you will find a couple of common themes.
First, for these figures it is a bygone conclusion that elevated cholesterol (e.g. LDL, ApoB, or lipoprotein A) is the causative agent of atherosclerosis.
That is to say, it is the necessary and sufficient factor to cause atherosclerotic narrowing of blood vessels.
Every new development in the field of cardiology and lipidology is finding a new canary to blame the plaque on. First it cholesterol, then it was LDL cholesterol, now its ApoB (a part of the LDL cholesterol particle) and Lipoprotein(a) (intrinsically tied to ApoB).
Until you ask some simple questions:
How come there are no atherosclerotic plaques in veins?
Why do they only form in arteries?
Why do veins grafted into arteries suddenly develop plaque?
Then, behold as the explanations commence.
The explanations which underscore the truth: that atherosclerotic plaques are not caused by cholesterol or any constituent thereof.
These two facts should slowly but surely shake your confidence in the cholesterol hypothesis:
Atheromas are by and large composed of breakdown products of the blood, not fat.
Atheromas do not form in veins.
Now we can start answering the question: what is causing atherosclerosis?
A Little History
The idea that dietary fat and blood cholesterol causes atherosclerosis is almost 100 years old now. Starting with the fraudulent work of Ancel Keys, the focus on inflammatory stress shifted toward dietary fats. The basic premise that if you eat saturated fats (like butter, lard, and animal meat) you would increase your chances of atherosclerotic heart disease.
Eventually, even the American Heart Association realized that dietary fat intake does not correlate with blood cholesterol levels.
People could now eat more than 1 egg per day. Hurray!
Then, the focus shifted toward blood cholesterol panels and serum cholesterol profiles. Including things like triglycerides, LDL, and HDL cholesterol.
LDL became the new boogieman. This paved the way for a new class of drugs called HMG-CoA reductase inhibitors - AKA, Statins.
If you don’t know why Statins suck, you can find out here:

Then, as more pointless and circular research was done…people started to realize a few things
LDL levels do not predict cardiac mortality as well as we’d like.
People who live the longest tend to have the highest cholesterol and LDL.
Contributors to atherosclerosis are far more varied than just blood cholesterol
Despite the public health and drug campaigns, more and more people still develop atherosclerotic heart disease…
The cholesterol levels of the oldest people on earth:

Invariably, the same old problem was repeatedly presenting itself as a thorn in the side of the a cholesterol hypothesis folk.
Inflammatory and biomechanical stress.
Thus, the formation of atheromas is a response to injury…aptly termd the “response-to-injury” hypothesis. The critics of this hypothesis will state that “there is no definitive evidence in vivo that endothelial injury or desquamation is necessary or sufficient for lesion formation.
In some ways, this statement is true.
In the presence of “injury,” without any blood products to lay down at the site of injury (to heal)…then no atheroma can form.
These critics will also point to evidence that atheromas can form in the absence of visible injury to the endothelium. They often mean tears, or ulcers, or “desquamation” which refers to loss of the lining of the vessel. Which is also true.
Because of these observations, they felt the need to introduce a new framework. Which is Lipid retention. That is…there are some low-grade, not consequentially definitive changes to the nature of the blood vessel…that increase permeability to lipids (and other blood products)…which allows lipids to move through the vessel lining…and maybe, sometimes form atheromas.
The term “leak” is used here. As if it’s purely by accident that the vessel is changing its conformation to allow fats and blood products to move through these walls, even in the absence of clear discontinuity (tear, ulcer, desquamation).
The ultimate explanatory framework they have come up with is the Response-to-Retention model. Their conclusion is:
“…subendothelial retention of atherogenic lipoproteins as the central pathogenic process in atherogenesis.”
In their conclusion they state:
“Although atherosclerosis is a complex and multifactorial process, we conclude that there exists a key pathogenic event, namely, lipoprotein retention, that is both necessary and sufficient to provoke lesion initiation in an otherwise-normal artery.”
To translate, they conclude that “retention” is both necessary and sufficient to explain the presence of an atheromatous lesion, in an “otherwise-normal” artery.
Retention leads to retention. Absolute genius.
What they don’t discuss in their article…as almost all articles on this topic that I have encountered is the following.
In all the in vivo studies they cite, they find that these lipid plaques form in 2 specific locations:
Only in arteries.
In specific locations in the artery.
Number 2 is where there is some sleight-of-hand used to obscure the truth.
All these studies discussing the Response-to-Retention model cite the work of Schwenke and Carew, who demonstrated the following:
accumulation of atherogenic lipoproteins within the arterial wall is focally concentrated at certain sites
The field has since termed these sites atheroma “prone” and other locations where atheromas do not form they termed atheroma “resistant.”
That’s it. That’s how they brush this conundrum aside, and why most don’t notice.
This is the part nobody talks about.
If the rate of lipid entry into the prone vs resistant sites is the same…then how are the lipids the problem?
Do you see?
Lipids are needed by all cells of the body…at all times.
So if the same amount is moving into the atheroma prone and atheroma resistant locations…then, why is one retaining the lipid but not the other?
Clearly, the causative agent is not the lipoprotein itself.
The real question is: why is one site prone and one site resistant?
Ultimately, the answer lies in a combination of electrodynamics, circulatory stress, systemic inflammation, and Poiseuille’s Law.
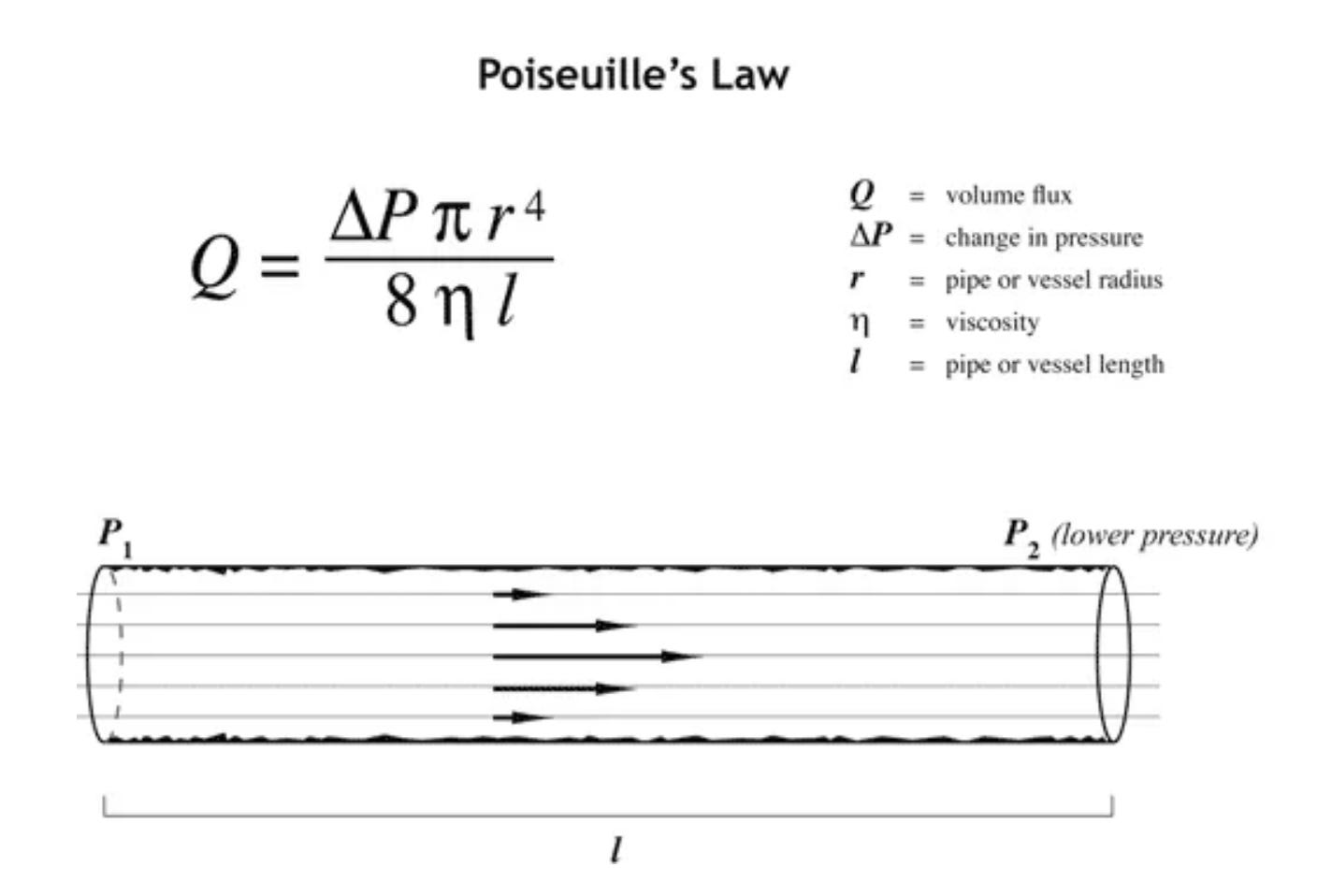
The modifiable factors in this Law that are relevant to our physiology are:
Viscosity of blood
Change in pressure
Vessel diameter
Closely related to all three of these factors, especially viscosity are the contours of our circulatory system, and the charge separation between blood vessel wall and the viscous/non-newtownian blood contained within.
Digging Into The Real Factors
In the near future, I will be hosting a Seminar and Q&A on the nature of circulation, circulatory stress, and all of its consequences including blood pressure, brain aneurysms and atherosclerosis.
If you want the opportunity to partake in the Seminar, submit questions, or just watch segments of the Seminar, hit subscribe and you’ll be among the first to know!
The amount of motivated reasoning on this topic is only topped by vaccinology.
They just can't let of of the notion that cholesterol is not the cause.
Or a "causal factor"
Or a "risk factor"
Or "strongly correlated."
It's never ending.
I have been waiting a long time for you to tackle this issue head on. I have very high LDL cholesterol but excellent triglycerides, HDL and lipoprotein (a) and a Coronary Artery Calcium score of 0 at >55 years old. BMI around 22. Fit, active and low carb diet. I fit the profile of what has recently been termed a "lean mass hyper responder". Nevertheless, my PCP and cardiologist insist on a statin because of their hyperfocus on my LDL cholesterol. It seems mainstream medicine is still wedded to the LDL-is-always-bad hypothesis. However, researchers such as Dave Feldman, Nick Norwitz and others, and now hopefully you, are poking holes in that theory. I look forward to reading more from you about this topic.